Understanding photoemission time delays using molecular frame photoelectron angular distributions
Written by: Jennifer Joseph
April 25, 2018
Probing the motion of electrons on the attosecond (10-18 s) time scale with the help of advanced light sources is a milestone in the research domain of light-matter interaction. By focusing an intense femtosecond (10-15s) laser pulse in a gas medium (mostly noble gases), it is possible to generate high order odd-harmonics of the fundamental (driving) laser field in a non-linear process. This technique, known as the high harmonic generation (HHG), produces a train of attosecond pulses. Having a pulse duration in the attosecond regime and frequencies extending up to extreme ultraviolet (XUV) energy region, these pulses are ideal for observing electron dynamics induced by photoionization. With this advanced experimental technique, time-resolved studies on electron correlation effects, observation of resonances, autoionizing states and photoemission delays in atoms, molecules and solids were realized.
With an attosecond pulse train (APT) corresponding to a comb of high-order harmonics in the XUV region as a pump and a weak infrared (IR) dressing laser as a probe, a sub-femtosecond time-resolved spectroscopic methodology known to the ultrafast community as the RABBITT technique (Reconstruction of Attosecond Beatings by Interference of Two photon Transitions) 1,2 was developed. In the RABBITT scheme, the XUV attosecond pulse ionizes the target gas and generates photoelectrons. In the absence of the dressing IR field, the photoelectron spectrogram consists of electrons peaks at energies (Ekin = EHarmonics - Ionization potential (IP)) corresponding to the harmonic peaks in the attosecond pulse train (odd harmonics in case of HHG, EHarmonics = (2q+1) ħωIR, where q is an integer). With a weak IR dressing laser delayed by time, τ, the emitted electrons interact with the laser field and undergo free-free transitions. This, in turn, produces satellite bands (at energies ESB = 2q ħωIR) between the harmonic peaks called ‘Sidebands’. As illustrated in Fig. 1, for the ionization of Ar from 3s and 3p shells, sidebands, at photoelectron energies Ekin corresponding to ESB = (2q) ħωIR, are formed as a result of interferences between 1) absorption of an IR photon by a photoelectron emitted by the 2q-1 harmonics, 2) stimulated emission of an IR photon by the photoelectron emitted by 2q+1 harmonics in the presence of the IR field. Hence, the same final state can be reached by two different coherent pathways, which leads to oscillations in the intensity due to interference. The sideband intensity is given as 3,4

where τ is the delay between the XUV and IR pulses, Δϕ2q is the phase difference associated with consecutive odd harmonics, and Δϕ1 is an intrinsic phase related to ionization of the target gas (atoms/molecules). The RABBITT method then pertains to energy resolved photoelectron spectroscopy and relies on the dependence of the intensity of the sidebands as a function of the delay between the XUV and IR dressing field. It was first performed for characterizing the XUV pulses through the determination of the relative phases of the harmonics, Δϕ2q. These experiments further provide an insight into the wavepacket dynamics associated with the studied photoionization process through the determination of the ΔϕI phases, assuming that the harmonic phases can be retrieved or subtracted. In one of the first RABBITT experiments in molecules, Mairesse and co-workers 5 obtained the amplitude and the phase of the two-colour two-photon ionization transition into different vibrational levels of the cationic ground state for nitrogen molecules. The observed phase shift was related to the Hopfield Rydberg state in the ionization continuum. In this article, we will be discussing the application of the RABBITT technique to extract time delays associated with photoionization. We report recent results obtained in this domain and present our project in this field for photoionization of molecules.
In equation (1), ΔϕI is the phase difference between the amplitudes related to the ionization of the target gas (atoms/molecules) through the two interfering paths, and is associated with the time delay in the temporal domain. The temporal delay is given by
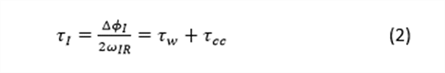
where τw defines the Wigner time delay 6 and τcc the continuum-continuum delay, which quantifies the interaction of the photoelectron in the continuum with the probe IR field. Wigner in 19556 first predicted the existence of a short time delay due to the electron scattering process in photoionization known as the Wigner time delay, which is defined as,

Here, η is the total scattering phase accumulated as a result of the interaction between the outgoing electron wave packet and the scattering potential, and ϵ is the energy of the photoelectron. It was only after the development of attosecond science that the first experiments were reported on the experimental proof of photoionization delays in atoms7,8.
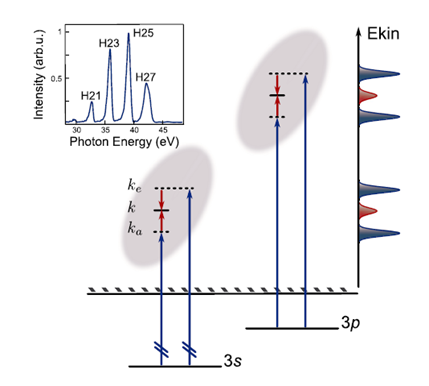
Figure 1. Ionizatoin from 3s and 3p orbitals of Argon and the observed photoelectron spectra. (Figure reprinted from reference Klunder et al., 2011)
The first experiments for studying time delays were preformed in rare gas targets, where relative photoemission delays were studied for the emission from two different states (orbitals) of the atom 7,8. As an example, in Argon (Fig. 1 and 2a, b), after two photon transitions using the RABBITT technique, a spectrogram is obtained where the electro peaks (Ekin) are shown at photon energies corresponding to Harmonic 21(21ħωIR), H23 and H25, and sidebands are shown at energies corresponding to 22ħωIR (SB22), 24ħωIR (SB24) and 26ħωIR (SB26).7 With this RABBITT spectrogram, oscillations in the SB intensity are measured as a function of XUV-IR delay, and the relative photoemission time delays are extracted. It is observed that, at sideband 22, (Fig. 2c) there is a delay of 40as between the photoemission from the 3s and 3p shells of argon. With these relative measurements (τI (3s) - τI (3p) of the two photoelectron wave packets, Wigner time delays are obtained.
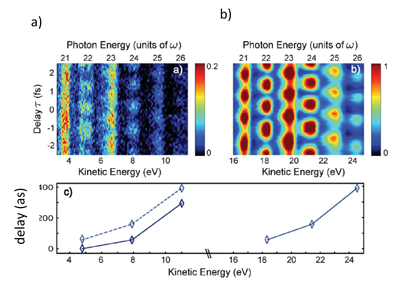
Figure 2. Photoelectron spectrogram obtained from RABBITT for the photoionization from 3s and 3p shells Argon and the change in sideband intensity with respect to the delay between pump and probe pulse. (Figure reprinted from reference Klunder et al., 2011).
Recent attosecond metrology experiments were performed in molecules at ETH Zurich following the same principle. They reveal that there is a photoionization time delay reaching 160as associated with the emission from two different electronic states ((τ(A+) - τI (X+)) in the case of N2O and 50as in H2O in the energy range of 20 to 40 eV9. Here, the longer time delays correspond to the shape resonance associated with valence shell ionization of N2O.
Another remarkable development in the photoionization studies in the time domain is the measurement of angle-resolved time delays,10–12 mostly developed for rare gases up to now. Here we obtain temporal information on the emission of electrons which are emitted from an initial state, but at different emission angles relative to the common polarization axis of XUV and IR. After absorption of one XUV photon, the electron in the p state can access two different final states, i.e either s state or d state (p→s/d). The interference between these two partial waves in the final electronic state gives rise to an angular dependence in the photoelectron emission11 and thus leads to an angle dependent Wigner time delay. Additional contributions to the angular dependence of the atomic emission delay may also arise from the continuum-continuum transition corresponding to the absorption of the IR photon13. Recent measurements by Cirelli et al. (2018)10 show how atomic delays are affected by the correlation dynamics associated with autoionizing resonances measured in Argon. A strong dependence of the emission delays with respect to the emission angles is observed near autoionizing resonances.
Studying angularly resolved time delays in photoionization of molecules, which involves electron scattering in an anisotropic potential, is more complex and challenging. Electron wavepacket experiencing an anisotropic molecular potential develop a scattering phase which is dependent on the emission angle in the molecular frame (MF), so the complete picture of the photoionization process and the associated dynamics in a molecule is better understood by studying the angular distribution of photoelectrons in the molecular frame. Time resolved molecular frame photoelectron angular distributions (MFPADs) are one of the most sensitive tools to access the complete photoionization experiment in this scenario.
Experimental studies on photoemission delays in molecules with the outlook on angular dependence are in their early stages. Few theoretical formalisms on these time delays in molecules already exist (one photon 14 and two photon cases 15), which take into consideration the angular dependence of the photoelectron emission and the corresponding time delays. In 2016, using the RABBITT technique and coincidence momentum spectroscopy, so-called stereo Wigner time delays, in CO were studied16. The asymmetry in the photoelectron emission from C and O ends of the molecule and the corresponding delays were measured, for an orientation of the molecule parallel to the common polarization axis of XUV and IR radiation. Stereo Wigner time delay (SWTD) Δτω is the time required for the photoelectron emission from either side of the molecule. In the case of CO, Δτω quantifies the photoemission delay associated with electrons escaping the anisotropic molecular ionic potential along the molecular axis at emission angles about 0° and 180° corresponding to the C and O ends of the molecule, respectively. With this relative measurement, it was possible to extract SWTD eliminating all other time delay contributions (τcc and τ2q). Future perspectives on this topic to measure the delays in all possible emission angles are challenging, but at the same time promising. These angle resolved time delay measurements will guide us in exploring complete photoionization dynamics in the temporal domain.
Our group at ISMO, CNRS-University of Paris Sud, investigates molecular photoionization in diatomic and small polyatomic molecules by measuring MFPADs using electron-ion coincidence 3D momentum spectroscopy. Studies on dissociative photoionization of small gas phase molecules using well characterized radiation sources (synchrotron radiation and attosecond laser sources) have already been performed in the group, which helped in understanding the inner-valence photoionization dynamics of small molecules (O2, NO, CO, NO2).17–19 In particular for the study of photoionization dynamics in NO, photoionization into the dominant state (c3Π) of NO in the inner valence region was explored using synchrotron radiation source20. Energy resolved MFPADs were recorded after identifying the process in the electron-ion energy correlation diagram known as KECD (Kinetic energy correlation diagram). Furthermore, the complete polarization state of the incoming radiation was extracted using molecular polarimetry techniques and compared to the data obtained using an in situ insertable VUV optical polarimeter installed at DESIRS beamline, SOLEIL21. This energy resolved spectroscopic measurements will provide a reference for the time resolved experiments.
With the motivation of understanding ultrafast photoionization dynamics, experiments were performed on NO using APT corresponding to harmonics H15 to H25 at PLFA laser facility (CEA-SLIC).22 They demonstrated the feasibility of measuring complete MFPADs based on coincidence experiments with a HHG radiation source at 1kHz repetition rate.
Dominant processes in the KECD corresponding to photoionization into the (c3Π) state of NO+ induced by the comb of harmonics are resolved as seen in Fig. 3a. Corresponding MFPADs are obtained and are in good agreement with the synchrotron measurements and multi-channel Schwinger configuration interaction (MCSCI) calculations20,22. As seen in Fig. 3b across the shape resonance region, there are differences in the photoelectron emission from N and O ends, owing to changes in the anisotropic potential and photoionization dynamics. As discussed before, in the time domain more information about the energy dependence of the scattering phases will be captured by the measurements of the photoemission time delays. With this motivation, our next goal is to obtain photoemission delays in NO across this shape resonance region by recording time resolved MFPADS. This work is in progress using electron-ion coincidence momentum spectroscopy on the FAB10 beamline (20 W, 2 mJ/10 kHz), 24 fs (15 fs in 2018)) at the ATTOLab attosecond laser facility (L’Orme des merisiers, CEA).
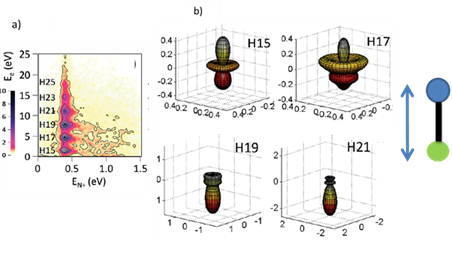
Figure 3. a) Kinetic energy correlation diagram showing photoionization into the c3Π state of NO+ using an APT containing harmonics H15 to H25. b) Evolution of the MFPADs for photoionization into the c3Π state of NO+ for a molecular axis orientated parallel to the incoming radiation polarization. Reproduced from ref22 with permission of The Royal Society of Chemistry.
Personal Experience as an Early Stage Researcher
It is my privilege to be a part of Horizon 2020 ASPIRE ITN framework. My journey as an ESR to date is one of the best experiences a young researcher could get in her career. As my colleagues have already mentioned, the opportunities we get through the training from the host institution (ISMO, CNRS-University of Paris-Sud), secondments, meetings (journal clubs, ASPIRE meetings) and conferences are changing our perspectives about the scientific world. It is a window to know the broad spectrum of the research carried out in leading groups across the world in the area of atomic and molecular photoionization.
References
1. Muller, H. G. Reconstruction of attosecond harmonic beating by interference of two-photon transitions. Appl. Phys. B 74, s17–s21 (2002).
2. Paul P.M., E S Toma, P Breger, G Mullot, F Auge, Ph Balcou, H G Muller, P Agostini. Science 292, 1689 (2001)
3. Dahlström, J. M., L’Huillier, A. & Maquet, A. Introduction to attosecond delays in photoionization. J. Phys. B At. Mol. Opt. Phys. 45, 183001 (2012).
4. Nisoli, M., Decleva, P., Calegari, F., Palacios, A. & Martin, F. Attosecond Electron Dynamics in Molecules. Chem. Rev. (2017). doi:10.1021/acs.chemrev.6b00453
5. Haessler, S. et al. Phase-resolved attosecond near-threshold photoionization of molecular nitrogen. Phys. Rev. A 80, 011404 (2009).
6. Wigner, E. P. Lower limit for the energy derivative of the scattering phase shift. Phys. Rev. 98, 145 (1955).
7. Klünder, K. et al. Probing Single-Photon Ionization on the Attosecond Time Scale. Phys. Rev. Lett. 106, (2011).
8. Schultze, M. et al. Delay in photoemission. science 328, 1658–1662 (2010).
9. Huppert, M., Jordan, I., Baykusheva, D., von Conta, A. & Wörner, H. J. Attosecond Delays in Molecular Photoionization. Phys. Rev. Lett. 117, (2016).
10. Cirelli, C. et al. Anisotropic photoemission time delays close to a Fano resonance. Nat. Commun. 9, (2018).
11. Heuser, S. et al. Angular dependence of photoemission time delay in helium. Phys. Rev. A 94, (2016).
12. Picard, Y. J. et al. Attosecond evolution of energy- and angle-resolved photoemission spectra in two-color (XUV + IR) ionization of rare gases. Phys. Rev. A 89, 031401 (2014).
13. Ivanov, I. A. & Kheifets, A. S. Angle-dependent time delay in two-color XUV+IR photoemission of He and Ne. Phys. Rev. A 96, (2017).
14. Hockett, P., Frumker, E., Villeneuve, D. M. & Corkum, P. B. Time delay in molecular photoionization. J. Phys. B At. Mol. Opt. Phys. 49, 095602 (2016).
15. Baykusheva, D. & Wörner, H. J. Theory of attosecond delays in molecular photoionization. J. Chem. Phys. 146, 124306 (2017).
16. Cattaneo, L., Vos, J., Lucchini, M., Cirelli, C. & Keller, U. Asymmetric Wigner time delay in CO photoionization. in International Conference on Ultrafast Phenomena UM2B–3 (Optical Society of America, 2016).
17. Lafosse, A. et al. Vector correlations in dissociative photoionization of O2 in the 20–28 eV range. I. Electron-ion kinetic energy correlations. J. Chem. Phys. 114, 6605–6617 (2001).
18. Lebech, M., Houver, J. C. & Dowek, D. Valence and inner-valence shell dissociative photoionization of CO in the 26–33 eV range. I. Ion-electron kinetic energy correlation and laboratory frame photoemission. J. Chem. Phys. 130, 194307 (2009).
19. Marggi Poullain, S. et al. The role of Rydberg states in photoionization of NO2 and (NO+, O−) ion pair formation induced by one VUV photon. J. Chem. Phys. 139, 044311 (2013).
20. Lebech, M. et al. Complete description of linear molecule photoionization achieved by vector correlations using the light of a single circular polarization. J. Chem. Phys. 118, 9653–9663 (2003).
21. Veyrinas, K. et al. Complete determination of the state of elliptically polarized light by electron-ion vector correlations. Phys. Rev. A 88, 063411 (2013).
22. Veyrinas, K. et al. Molecular frame photoemission by a comb of elliptical high-order harmonics: a sensitive probe of both photodynamics and harmonic complete polarization state. Faraday Discuss. 194, 161–183 (2016).