Computational Modelling and Informatics
Our key interest: molecular structure, flexibility and recognition
At the heart of most of our research activities is the development and application of state-of-the-art molecular simulation methods to the study of the structure, dynamics, and recognition properties of complex molecular systems. Most commonly these are biomolecular systems (proteins and/or nucleic acids), but we also apply the methods in other areas (e.g. drug-polymer complexes). Some of our research is quite “blue skies”, related to understanding the fundamental principles behind the relationship between molecular structure and dynamical behaviour, and while other projects are collaborative activities with experimental groups aimed at discovering and developing new drugs. As well as using existing state-of-the-art simulation methods, an important part of our work is concerned with software development.
Structure, dynamics and recognition properties of G-protein coupled receptors
In collaboration with medicinal chemists and pharmacologists, we study a range of GPCRs of pharmaceutical interest. We are particularly concerned with the analysis of receptor dynamics: how this mediates signal transduction, and how this can be affected by both ligand binding and the physical environment of the receptor.
Software Development: new simulation and analysis methods
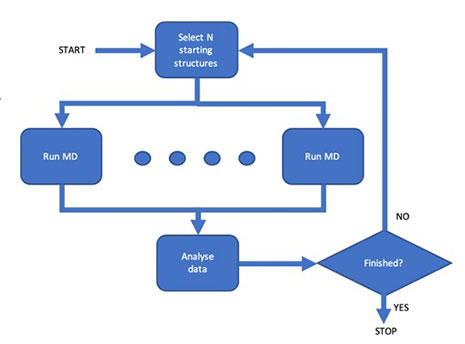
There is a constant drive for faster and more efficient molecular simulation methods, but also as a result a pressing need for effective methods to extract the maximum amount of useful information from the enormous amounts of raw data such simulations produce. We develop software to make it easier to use High Performance and Cloud computing resources for large-scale simulations, and for complex workflows.
Drug-polymer interactions and self-assembly
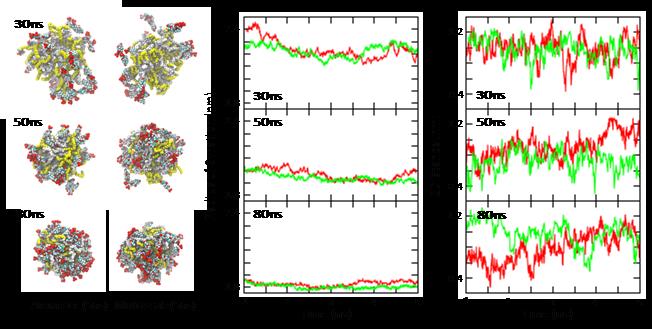
We use techniques that were first applied in the area of biomolecular simulation to study other types of large complex systems, particularly drug-polymer complexes. Examples include the simulation of polymer-drug nanoparticle self-assembly, and the evaluation of theoretical methods to predict polymer-drug compatibility.